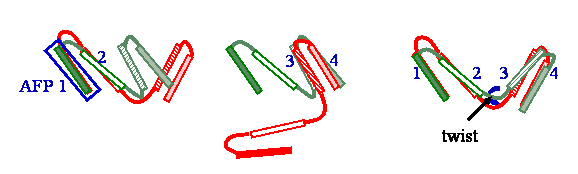
- AFP (Aligned Fragment Pair)
Given two protein structures, an AFP is defined as a match of two fragments, one from
each structure.
Each AFP defines the transformation (rotation and translation) needed to superimpose the
fragments included in it.
The figure below shows two AFPs, which define two different transformations of the input
structures.

- Twist
As shown in the schematic example below, the green structure has to be rearranged
by introducing a twist at the hinge (pointed to by the arrow) so that the green and
red structures can be better aligned
(i.e., including 1-4 AFPs, instead of only two, either 1-2 or
3-4).
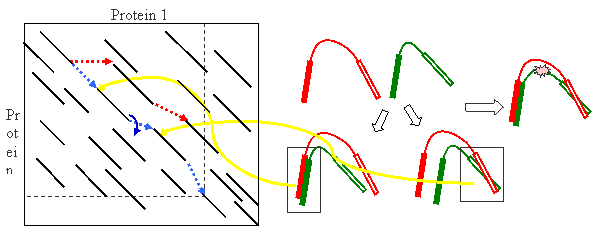
- FATCAT (chaining) score
In FATCAT, flexible structure alignment is formulated as an AFP
chaining process
(the path connected by blue dotted lines in the example alignment graph below)
allowing at most t twists (here t=5).
Dynamic programming is used in the chaining process (as shown in the figure below).
If we denote S(k) as the best score of paths ending at the AFP k, then it can be
calculated
from the best score of paths ending at previous AFPs that can be connected to the
k-th AFP, subject to the constraints.
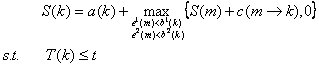
where a(k) is the score of AFP k itself, determined by its RMSD and length with long
AFPs rewarded and large RMSDs penalized; 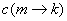 is the score of introducing a connection between AFP m and AFP k, defined by a
function of the compatibility of the
AFPs and the mis-matched regions and/or gaps created by the connection of the two
AFPs;
T(k) is the number of twists required to connect the chain of AFPs leading up to
S(k).
is the score of introducing a connection between AFP m and AFP k, defined by a
function of the compatibility of the
AFPs and the mis-matched regions and/or gaps created by the connection of the two
AFPs;
T(k) is the number of twists required to connect the chain of AFPs leading up to
S(k).
The FATCAT (chaining) score is the best of all S(k) in the alignment graph.
- P-value
A P-value is used in FATCAT to evaluate the significance of the detected structural
similarities detected. It is
the probability of observing a similarity score higher than the one currently
obtained, for unrelated structures.
It is calculated based on the observation that the
FATCAT similarity score between two unrelated structures follows the extreme value
distribution.
The FATCAT similarity score incorporates the FATCAT chaining
score, the
RMSD of the resulting superposition, the number of equivalent positions in the
alignment and the number of twists.
The FATCAT similarity score is computed as
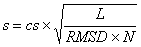
where cs is the FATCAT chaining score; L is the number of
equivalent positions in the alignment;
RMSD is the overall RMSD between two the structures when one structure is rearranged
at the positions
where twists are detected by FATCAT; N is the number of blocks in the alignment
(number of twists + 1).
The p-value of s is then computed as

where the location and the scale parameter of the extreme value distribution of
FATCAT similarity scores of
unrelated structures were determined by empirical simulation. For more details about
FATCAT significance calculation and
examples illustrating its possible biological interpretations, please see the
original
publication.
- align-len
The length of the alignment (including gaps)
- opt-len
The number of equivalent positions in the alignment
opt-len = align-len - gaps
- opt-rmsd
The root mean square deviation (RMSD) of aligned Cα atoms of the input
structures, with one input structure rearranged
if flexibility is detected (by introducing twists in the alignment).
- chain-rmsd
The root mean square deviation (RMSD) of aligned Cα atoms of the input structures,
without structural rearrangement (twists)
even if structural flexibility is detected in the alignment. It means that in the cases
with flexibility (when twists are introduced to get the alignment),
the value of chain-rmsd could be very high (because flexible alignment is longer than
rigid alignment would be).
The comparison of chain-rmsd and opt-rmsd makes it possible to asses how signifcantly
the introduced conformational flexibility
improved the alignment.
- transformed 'complete' structure
In FATCAT, only the Cα atoms of a single chain from two input structures are
aligned. If users choose to download a "complete" structure,
the server applies the transformation matrix to the "original" structure file (which may
contain coordinates of other chains, ligands etc)
and returns the transformed file. This output file is only available for FATCAT pairwise
alignment calculated with the rigid option.